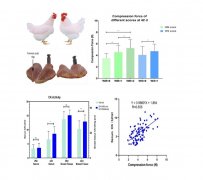
兽用化学药物稳定性研究技术指导原则
一、概述
兽用化学药物的稳定性是指原料药及制剂保持其物理、化学、生物学和微生物学的性质,通过对原料药和制剂在不同条件(如温度、湿度、光照等)下稳定性的研究,掌握兽药质量随时间变化的规律,为兽药的生产、包装、运输、贮存条件和有效期的确定提供依据,以确保临床用药的安全性和临床疗效。
稳定性研究是兽药质量控制研究的主要内容之一,与兽药质量研究和质量标准的建立紧密相关。稳定性研究具有阶段性特点,贯穿兽药研究与开发全的过程,一般始于兽药的临床前研究,在兽药临床研究期间和上市后还应继续进行稳定性研究。
本文为一般性原则,具体的试验设计和评价应遵循具体问题具体分析的原则。
二、稳定性研究的设计要求
稳定性研究的设计应根据不同的研究目的,结合原料药的理化性质、剂型的特点和具体的处方及工艺条件进行。
稳定性研究包括影响因素试验、加速试验和长期试验。原料药需进行影响因素试验、加速试验和长期试验;制剂需进行影响因素试验中的光照试验、加速试验和长期试验。
(一)样品的批次和规模
影响因素试验采用1批样品进行,加速试验和长期试验均采用3批样品进行。稳定性研究应采用一定规模生产的样品,以能够代表规模生产条件下的产品质量。原料药的合成工艺路线、方法、步骤应与最终生产规模一致;药物制剂的处方、制备工艺、设备也应与最终生产规模一致。
稳定性研究中,原料药的批量应达到中试规模的要求;质量应既代表临床前研究的质量,又能代表临床研究以及规模生产时的质量。口服固体制剂如片剂、胶囊剂应不少于10000个制剂单位,预混剂、可溶性粉剂不少于200公斤。大体积包装的制剂(如静脉输液等)批量至少应为各项试验所需总量的10倍。特殊品种、特殊剂型所需数量,视具体情况而定。制剂质量应能代表规模生产时的质量。
(二)包装及放置条件
稳定性试验要求在一定的温度、湿度、光照条件下进行,应充分考虑到药物在贮存、运输及使用过程中可能遇到的环境因素。
原料药的加速试验和长期试验所用包装应采用模拟小包装,所用材料和封装条件应与大包装一致。药物制剂应在影响因素试验结果的基础上选择合适的包装,加速试验和长期试验中的包装应与拟上市包装一致。稳定性研究中所用设备应能较好地对各项试验条件要求的环境参数进行控制和监测。
(三)考察时间点
由于稳定性研究的目的是考察兽药质量随时间变化的规律,因此研究中需要设置多个时间点考察样品的质量变化。
考察时间点应根据兽药的理化性质、稳定性趋势设置。如长期试验中,总体考察时间应涵盖所预期的有效期,中间取样点的设置要考虑兽药的稳定性特点和剂型特点。对某些环境因素敏感的兽药,应适当增加考察时间点。
(四)考察项目
稳定性研究的考察项目应选择在兽药保存期间易于变化,并有可能影响兽药的质量、安全性和有效性的项目,具体品种的考察项目设置应结合兽药的特性进行。
稳定性研究中如样品发生了显著变化,试验应中止,改变试验条件后再进行。
1. 原料药的“显著变化”应包括:
1.1 性状,如颜色、熔点、溶解度、比旋度超出拟定的标准规定,晶型、水分等变化超出拟定的标准规定。
1.2 含量测定超出拟定的标准规定。
1.3 有关物质,如降解产物、异构体的变化等超出拟定的标准规定。
1.4 结晶水发生变化。
2. 药物制剂的“显著变化”包括:
2.1 含量测定中发生5%的变化;或者不能达到生物学或者免疫学的效价指标。
2.2 任何一个降解产物超出拟定的标准规定。
2.3 性状、物理性质以及特殊制剂的功能性试验(如颜色、再混悬能力、结块、硬度等)超出拟定的标准规定。
2.4 pH 值超出拟定的标准规定;
2.5 制剂溶出度或释放度超出拟定的标准规定。
(五)分析方法
评价指标所采用的分析方法应经过充分的验证,能满足研究的要求,具有一定的专属性、准确性、灵敏度、重现性等。
三、稳定性研究的试验方法
根据研究目的和条件的不同,稳定性研究内容分为影响因素试验、加速试验、长期试验等。
(一)影响因素试验
在剧烈条件下进行影响因素试验,目的是了解影响稳定性的因素及可能的降解途径和降解产物,为制剂工艺筛选、包装材料和容器的选择、贮存条件的确定等提供依据。同时为加速试验和长期试验应采用的温度和湿度等条件提供依据,还可为分析方法的选择提供依据。
影响因素试验包括高温、高湿、强光照射试验。试验时将原料药供试品置适宜的容器中(如称量瓶或培养皿),摊成≤5mm厚的薄层,疏松原料药摊成≤10mm厚的薄层进行试验。对于制剂产品,则将最小包装的产品,单层排列置试验条件下进行强光照射试验。如试验结果不明确,应加试两个批号的样品。对于某些制剂,如软膏、注射液,应提供低温条件下的试验数据(如注射剂的冻融试验),以确保在低温条件下的稳定性。对于需要溶解或者稀释后使用的兽药,如注射用粉针剂、溶液、消毒剂等,还应考察临床使用条件下的稳定性。
1. 高温试验
供试品置密封洁净容器中,在60℃条件下放置10天,于第5天和第10天取样,检测有关指标。如供试品发生显著变化(如制剂含量下降5%),则在40℃下同法进行试验。如60℃无显著变化,则不必进行40℃试验。
2. 高湿试验
供试品置恒湿密闭容器中,于25℃、相对湿度(RH)90%±5%条件下放置10天,在第5天和第10天取样检测。检测项目应包括吸湿增重项(准确称取试验前后供试品的重量)。若吸湿增重5%以上,则应在25℃、RH75±5%下同法进行试验;若吸湿增重5%以下,且其他考察项目符合要求,则不再进行此项试验。
恒湿条件可采用恒温恒湿箱或通过在密闭容器下部放置饱和盐溶液来实现。根据不同的湿度要求,选择NaCl饱和溶液(15.5~60℃,RH75%±1%)或KNO3饱和溶液(25℃,RH92.5%)。
3. 强光照射试验
供试品置装有日光灯的光照箱或其它适宜的光照容器内,于照度4500±500Lx条件下放置10天,在第5天和第10天取样检测。
以上为影响因素稳定性研究的一般要求。根据兽药的性质必要时可以设计其他试验,如考察pH值、氧、冷冻等因素对兽药稳定性的影响。
(二)加速试验
在超常条件下进行加速试验的目的是通过加快市售包装中兽药的化学或物理变化速度来考察兽药稳定性,对兽药在运输、保存过程中可能会遇到的短暂的超常条件下的稳定性进行模拟考察,并初步预测样品在规定的贮存条件下长期稳定性。加速试验应取拟上市包装的3批样品进行,温度需比长期试验放置温度至少高15℃。一般可选择40±2℃、RH75%±5%条件下,进行6个月试验。在试验期间第0、1、2、3、6个月末取样检测考察项目。如在6个月内供试品经检测不符合质量标准要求或发生显著变化,则应在中间条件30℃±2℃、RH65%±5%同法进行6个月试验。
对采用不可透过性包装的含有水性介质的制剂,如溶液剂、混悬剂、乳剂、注射液等的稳定性研究中可不要求相对湿度。对采用半通透性的容器包装的药物制剂,如多层共挤PVC软袋装注射液、塑料瓶装乳房注入剂等,加速试验应在40℃±2℃、RH20%±5%的条件下进行。
乳剂、混悬剂、软膏剂、糊剂、眼膏剂、栓剂、浇泼剂、外用溶液剂、泡腾片及泡腾颗粒等制剂宜直接采用30℃±2℃、RH65%±5%(Na
2Cr
2O
7饱和溶液)的条件进行试验。对温度敏感药物(预计需在冰箱中4~8℃冷藏保存)的加速试验可在25℃±2℃、RH60%±5%条件下同法进行。需要冷冻保存的兽药可不进行加速试验。
(三)长期试验
长期试验的目的是考察兽药在运输、贮存、使用过程中的稳定性,能更直接地反映兽药稳定性特征,是确定有效期和贮存条件的最终依据。
应取拟上市包装3批样品在25±2℃、RH60%±10%条件进行试验,取样时间点在第一年一般为每3个月一次,第二年为每6个月一次,以后每年一次。对温度敏感药物的长期试验可在6℃±2℃条件下进行试验,取样时间同上。
(四)兽药上市后的稳定性研究
兽药在注册阶段进行的稳定性研究,并不能够完全代表实际生产产品的稳定性,具有一定的局限性。采用实际条件下生产的产品进行的稳定性考察的结果,是确定上市兽药稳定性的最终依据。
在兽药获准生产上市后,应采用实际生产规模的兽药继续进行长期试验,必要时还应进行加速试验和影响因素试验。根据继续进行的稳定性研究的结果,对包装、贮存条件和有效期进行进一步的确认。
兽药在获得上市批准后,可能会因各种原因而申请对制备工艺、处方组成、规格、包装材料等进行变更,一般应进行相应的稳定性研究,以考察变更后兽药的稳定性趋势,并与变更前的稳定性研究资料进行对比,以评价变更的合理性。
四、稳定性研究结果的评价
兽药稳定性的评价是对稳定性研究中的各项试验,如影响因素试验、加速试验、长期试验中得到的兽药稳定性信息进行系统的分析和结果判断。
(一)贮存条件的确定
新药注册申请应综合影响因素试验、加速试验和长期试验的结果,同时结合兽药在流通过程中可能遇到的情况进行综合分析。选定的贮存条件应按照规范术语描述。
(二)包装材料/容器的确定
根据影响因素试验结果,初步确定包装材料和容器,结合加速试验和长期试验的稳定性研究的结果,进一步验证采用的包装材料和容器的合理性。
(三)有效期的确定
兽药的有效期应综合加速试验和长期试验的结果,进行适当的统计分析得到,最终有效期的一般以长期试验的结果来确定。
由于试验数据的分散性,一般应按95%可信限进行统计分析,得出合理的有效期。如3批统计分析结果差别较小,则取其平均值为有效期,如差别较大则取其最短的为有效期。若数据表明测定结果变化很小,表明兽药是很稳定的,则可以不做统计分析。
五、名词解释
有效期:系指一段时间内,上市包装兽药在规定的贮存条件下放置,其质量仍符合注册质量标准。
批次:按相同的生产工艺在一次生产过程中生产的一定数量的原料药或制剂,其质量具有均一性。
上市包装:上市销售兽药的内包装和其他层次包装的总称。
六、附录
(一)国际气候带
稳定性长期试验所采用的一般条件是根据国际气候带制定的。国际气候带将全球分为I、II、III、IV 四个气候带,具体条件见下表:
|
气候带 |
计算数据 |
推算数据 |
|
温度① |
MKT② |
湿度 |
温度 |
湿度 |
|
I 温带 |
20.0 |
20.0 |
42 |
21 |
45 |
|
II 地中海气候,亚热带 |
21.6 |
22.0 |
52 |
25 |
60 |
|
III 干热带 |
26.4 |
27.9 |
35 |
30 |
25 |
|
IV 湿热带 |
26.7 |
27.4 |
76 |
30 |
70 |
①记录温度;②平均热力学温度
温带主要有英国、北欧、加拿大、俄罗斯;亚热带有美国、日本、西欧(葡萄牙-希腊);干热带有伊朗、伊拉克、苏丹;湿热带有巴西、加纳、印度尼西亚、尼加拉瓜、菲律宾。在这四种气候带中,对于兽药的质量保证而言,条件最苛刻的是第四种气候带,即高温又高湿的环境。中国总体来说属于亚热带,推荐长期试验采用温度湿度条件为:25℃±2℃,60%RH±10%RH,与ICH所采用的条件基本一致。
(二)对半通透性容器的一些要求
对于包装在半通透性容器包装内的以水为溶剂的药物制剂,除对其常规项目进行考察外,还应对其可能发生的水的损失进行评价。此类兽药应能耐受低湿度环境。其加速试验应在较低的相对湿度下进行,推荐的试验条件如下:
加速:40±2℃/ RH 20%±5%
中等:30±2℃/ RH 40%±5%
长期:25±2℃/ RH 40%±10%
如果长期试验在25℃±2℃/40%RH±10%条件下进行,并且在加速试验的6个月中发生了除水分减失以外的显著变化,应进行中等条件下的附加试验。对于包装在半通透性容器包装内的药物产品,在经过25℃±2℃/RH40%±10%条件下平衡放置3个月后,与其初始值发生5%的水分减失被认为是显著变化。
(三)稳定性研究报告的一般内容
注册申请资料中研究数据应完整、可靠,资料整理应规范、清晰。稳定性研究部分的申报资料应包括以下内容:
1. 供试兽药的品名、规格、剂型、批号、生产者、原料药的来源、生产日期和试验开始时间。应明确给出稳定性考察中各个批次兽药的批产量。
2. 各稳定性试验的条件,如温度、光照强度、相对湿度、容器的开放/封闭、直立/颠倒放置、容器中干燥剂的使用等。应明确包装/密封系统的性状,如包材类型、形状和颜色等。
3. 稳定性研究中各考察项目的检测方法和指标的限度要求。
4. 在研究起始和试验中间的各个取样点获得的实际检测数据,应以表格的方式提交。
5. 检测结果和数据应如实填写申报,不宜采用“符合要求”等词简略表述。含量检测结果应该用每个制剂单位含有有效成分的量,如μg,mg,g 等表示,并给出其与初始值的百分比。如果在某个时间点进行了多次检测,应提供所有的检测结果及其相对标准偏差(RSD)。
6. 应对试验结果进行分析并得出初步的结论。
7. 不同的试验条件、不同的时间点取样检测得到的图谱均应附在稳定性试验报告之后。
(四)稳定性研究重点考察项目
主要剂型见附表,表中未列入的剂型的考察项目,见药典附录有关剂型项下的要求。
附表:原料药及药物制剂稳定性重点考察项目表
|
剂 型 |
稳定性重点考察项目 |
|
原料药 |
性状、熔点、含量、降解产物、以及根据药物性质选定的考察项目 |
|
片剂 |
性状、含量、降解产物、溶出度 |
|
胶囊剂 |
性状、内容物色泽、含量、降解产物、溶出度、水分 |
|
注射剂 |
色泽、含量、pH值、澄明度、降解产物、不溶性微粒、塑料容器胶塞等还应检查可抽提物 |
|
软膏剂 |
性状、含量、均匀性、降解产物(乳膏还应检查分层现象) |
|
眼膏剂 |
性状、含量、均匀性、粒度、降解产物、无菌。 |
|
滴眼剂 |
如为溶液,应考察:性状、澄明度、含量、pH值、降解产物、无菌*;如为混悬型,应考察:性状、沉降体积比、粒度、含量 |
|
溶液剂 |
性状、含量、色泽、澄清度、降解产物(外用溶液剂还应检查乳化稳定性) |
|
乳剂 |
性状、含量、分层测定、降解产物 |
|
混悬剂 |
性状、含量、沉降体积比、颗粒细度、降解产物 |
|
粉剂 |
性状、降解产物、含量、外观均匀度、干燥失重、含量均匀度(小规格) |
|
气雾剂 |
容器严密性、含量、降解产物、每揿次的释放量。 |
|
颗粒剂 |
性状、含量、降解产物、溶化性、释放度或溶出度 |
|
透皮溶液剂 |
性状、含量、降解产物、释放度 |
|
乳房注入剂 |
性状、含量、降解产物、无菌 |
|
栓剂 |
性状、含量、降解产物、熔变时限、溶出度或释放度 |
|
预混剂 |
性状、含量、降解产物、含量均匀度(小规格) |
注:降解产物应说明分解产物数目的变化及量的变化。